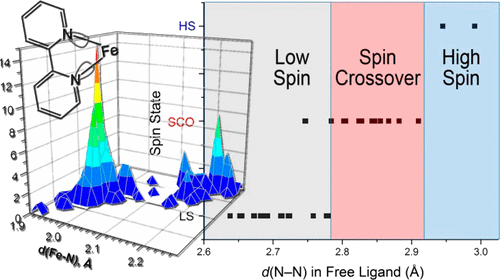
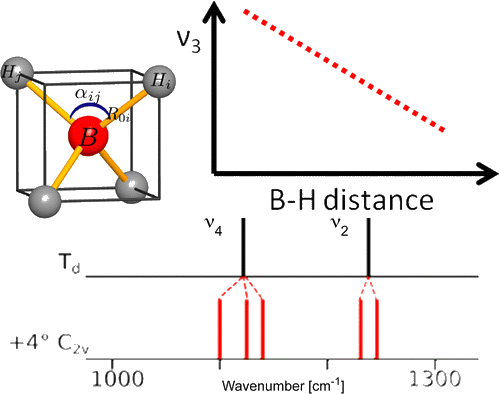
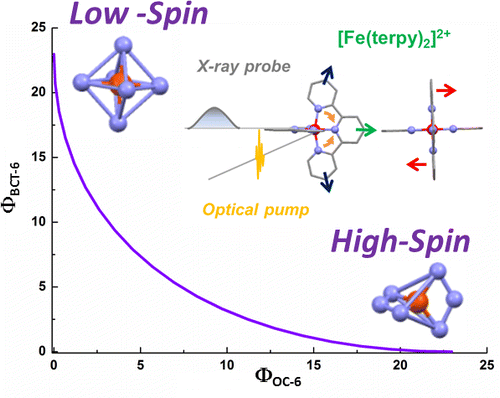
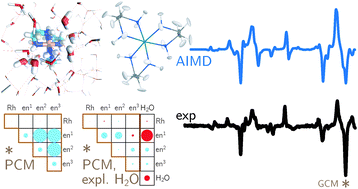
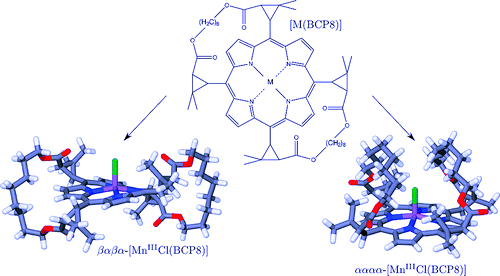
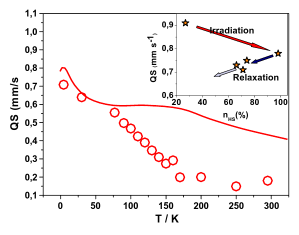
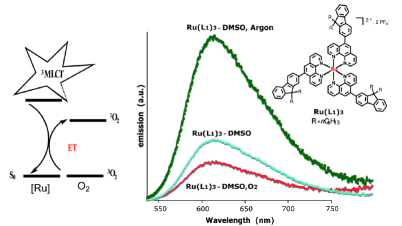
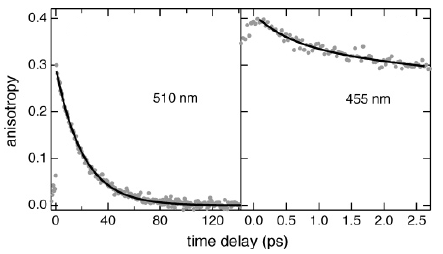
Magnetization measurements and variable temperature optical spectroscopy have been used to investigate, within the 4â300 K temperature range, the electronic structure of the reduced high-potential iron protein (HiPIP) from Chromatium vinosum and the model compounds (Cat)2[Fe4S4(SR)4], where RS- = 2,4,6-triisopropylphenylthiolate (1), 2,6-diphenylphenylthiolate (2), diphenylmethylthiolate (3), 2,4,6-triisopropylbenzylthiolate (4, 4â), 2,4,6-triphenylbenzylthiolate (5, 5â), 2,4,6-tri-tert-butylbenzylthiolate (6), and Cat+ = +NEt4 (1, 2, 3, 4â, 5â, 6), +PPh4 (4, 5). The newly synthesized 22-, 32-, 52-, and 62- complexes are, as 12- and 42-, excellent models of the reduced HiPIPs: they exhibit the [Fe4S4]3+/2+ redox couple, because of the presence of bulky ligands which stabilize the [Fe4S4]3+ oxidized core. Moreover, the presence of SCH2 groups in 42-, 52-, and 62-, as in the [Fe4S4] protein cores, makes them good biomimetic models of the HiPIPs. The X-ray structure of 2 is reported: it crystallizes in the orthorhombic space group Pcca with no imposed symmetry and a D2d-distorted geometry of the [Fe4S4]2+ core. Fit of the magnetization data of the reduced HiPIP and of the 1, 2, 3, 4, 5, and 6 compounds within the exchange and double exchange theoretical framework leads to exchange coupling parameters J = 261â397 cm-1. A firm determination of the double exchange parameters B or, equivalently, the transfer integrals β = 5B could not be achieved that way. The obtained |B| values remain however high, attesting thus to the strength of the spin-dependent electronic delocalization which is responsible for lowest lying electronic states being characterized by delocalized mixed-valence pairs of maximum spin 9/2. Electronic properties of these systems are then accounted for by the population of a diamagnetic ground level and excited paramagnetic triplet and quintet levels, which are respectively J and 3J above the ground level. Optical studies of 1, 2, 4â, 5â, and 6 but also of (NEt4)2[Fe4S4(SCH2C6H5)4] and the isomorph (NEt4)2[Fe4S4(S-t-Bu)4] and (NEt4)2[Fe4Se4(S-t-Bu)4] compounds reveal two absorption bands in the near infrared region, at 705â760 nm and 1270â1430 nm, which appear to be characteristic of valence-delocalized and ferromagnetically coupled [Fe2X2]+ (X = S, Se) units. The |B| and |β| values can be directly determined from the location at 10|B| of the low-energy band, and are respectively of 699â787 and 3497â3937 cm-1. Both absorption bands are also present in the 77 K spectrum of the reduced HiPIP, at 700 and 1040 nm (Cerdonio, M.; Wang, R.-H.; Rawlings, J.; Gray, H. B. J. Am. Chem. Soc. 1974, 96, 6534â6535). The blue shift of the low-energy band is attributed to the inequivalent environments of the Fe sites in the protein, rather than to an increase of |β| when going from the models to the HiPIP. The small differences observed in known geometries of [Fe4S4]2+ clusters, especially in the FeâFe distances, cannot probably lead to drastic changes in the direct FeâFe interactions (parameter β) responsible for the delocalization phenomenon. These differences are however magnetostructurally significant as shown by the 261â397 cm-1 range spanned by J. The cluster's geometry, hence the efficiency of the Feâμ3-SâFe superexchange pathways, is proposed to be controlled by the more or less tight fit of the cluster within the cavity provided by its environment. |